Fibrosis quística
Definición
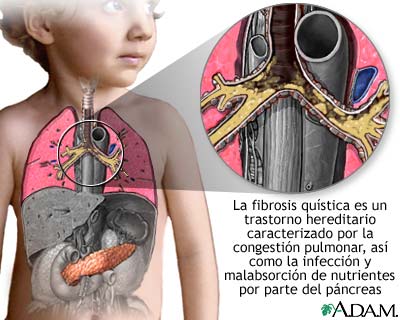
Es una enfermedad que provoca la acumulación de moco espeso y pegajoso en los pulmones, el tubo digestivo y otras áreas del cuerpo. Es uno de los tipos de enfermedad pulmonar crónica más común en niños y adultos jóvenes. Es una enfermedad potencialmente mortal.
Nombres alternativos
FQ
Causas
La fibrosis quística (FQ) es una enfermedad hereditaria. Es causada por un gen defectuoso que lleva al cuerpo a producir un líquido anormalmente espeso y pegajoso llamado moco. Este moco se acumula en las vías respiratorias de los pulmones y en el páncreas.
Esta acumulación de moco ocasiona infecciones pulmonares potencialmente mortales y serios problemas digestivos. Esta enfermedad también puede afectar las glándulas sudoríparas y el aparato reproductor masculino.
Muchas personas portan el gen de la FQ, pero no manifiestan ningún síntoma. Esto se debe a que una persona con esta enfermedad debe heredar 2 genes defectuosos, 1 de cada padre. Algunos estadounidenses tienen el gen de la FQ. La enfermedad es más frecuente entre aquellas personas descendientes de europeos del centro y norte.
A la mayoría de los niños con FQ se les diagnostica la enfermedad hacia los 2 años de edad, especialmente cuando se realiza un examen de detección en recién nacidos en todo Estado Unidos. Para un pequeño número, la enfermedad no se detecta hasta la edad de 18 años o más. Estos niños con frecuencia padecen una forma más leve de la enfermedad.
Síntomas
Los síntomas en los recién nacidos pueden incluir:
Retraso en el crecimiento - Incapacidad para aumentar de peso normalmente durante la niñez
- Ausencia de deposiciones durante las primeras 24 a 48 horas de vida
- Piel con sabor salado
Los síntomas relacionados con la función intestinal pueden incluir:
- Dolor abdominal a causa del estreñimiento grave
- Aumento de gases, meteorismo o un abdomen que parece hinchado (distendido)
- Náuseas e inapetencia
Heces pálidas o color arcilla , de olor fétido, que tienen moco o que flotan- Pérdida de peso
Los síntomas relacionados con los pulmones y los senos paranasales pueden incluir:
- Tos o aumento de la mucosidad en los senos paranasales o los pulmones
Fatiga - Congestión nasal causada por los pólipos nasales.
- Episodios recurrentes de
neumonía (los síntomas de neumonía en una persona con fibrosis quística abarcan fiebre, aumento de la tos y dificultad respiratoria, aumento de la mucosidad y pérdida del apetito) - Dolor o presión sinusal causados por infección o
pólipos
Los síntomas que se pueden notar posteriormente en la vida son:
- Esterilidad (en los hombres)
- Inflamación repetitiva del páncreas (
pancreatitis ) - Síntomas respiratorios
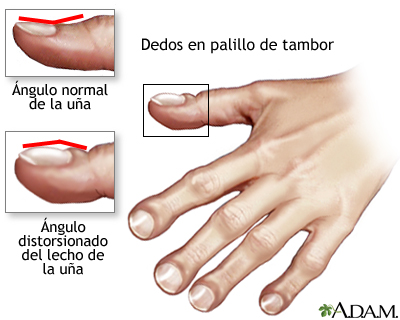
- Dedos malformados
Pruebas y exámenes
Se lleva a cabo un examen de sangre para ayudar a detectar la FQ. El examen busca variaciones en el gen de la FQ. Otros exámenes utilizados para diagnosticar la FQ incluyen:
- El examen del tripsinógeno inmunorreactivo (IRT, por sus siglas en inglés) es una prueba de detección estándar para FQ en recién nacidos. Un alto nivel de IRT sugiere una posible FQ y requiere exámenes adicionales.
- La
prueba de cloruro en el sudor es el examen diagnóstico estándar para la FQ. Un alto nivel de sal en el sudor de la persona es una señal de la enfermedad.
Otros exámenes para identificar problemas que pueden estar relacionados con la FQ incluyen:
Radiografía de tórax otomografía computarizada Examen de grasa fecal Pruebas de la función pulmonar - Medición de la función pancreática (elastasa pancreática en heces)
Examen de estimulación de secretina Tripsina y quimiotripsina en heces Tránsito esofagogastroduodenal - Cultivos pulmonares (obtenidos por esputo, broncoscopia o muestra faríngea)
Tratamiento
Un diagnóstico temprano de FQ y un plan de tratamiento pueden mejorar tanto la supervivencia como la calidad de vida. El control y vigilancia son muy importantes. Cuando sea posible, se deben recibir cuidados en clínicas con especialidad en fibrosis quística. Cuando los niños llegan a la adultez, deben transferirse a un centro especializado en fibrosis quística para adultos.
El tratamiento para los problemas pulmonares incluye:
- Antibióticos para prevenir y tratar infecciones sinusales y pulmonares. Se pueden tomar por vía oral o aplicarse por vía intravenosa o por medio de tratamientos respiratorios. Las personas con FQ pueden tomar antibióticos solo cuando sea necesario o todo el tiempo. Las dosis a menudo son más altas de lo normal.
- Medicamentos inhalados para ayudar a abrir las vías respiratorias.
- Otros medicamentos administrados por medio de un tratamiento respiratorio para diluir el moco y facilitar la expectoración son la terapia con la enzima DNAasa y las soluciones salinas altamente concentradas (solución salina hipertónica).
- Vacuna antigripal y
vacuna antineumocócica de polisacáridos (PPV, por sus siglas en inglés) anualmente (pregúntele a su proveedor de atención médica). - El
trasplante de pulmón es una opción en algunos casos. - Es posible que se necesite oxigenoterapia a medida que la enfermedad pulmonar empeore.
Los problemas pulmonares también se tratan con terapias para diluir el moco. Esto facilita su expectoración fuera de los pulmones.
Estos métodos incluyen:
- Actividad o ejercicio que lo llevan a respirar profundo
- Dispositivos que se usan durante el día para ayudar a despejar las vías respiratorias de las cantidades excesivas de moco
- Percusión manual del pecho (o fisioterapia del pecho), en la cual un miembro de la familia o un terapeuta dan palmadas suavemente sobre el pecho, la espalda o un área por debajo de los brazos de la persona
El tratamiento para problemas intestinales y
- Una dieta especial rica en proteínas y calorías para niños mayores y adultos
- Enzimas pancreáticas para ayudar a absorber grasas y proteínas, que se toman con cada comida
- Suplementos vitamínicos, sobre todo las vitaminas A, D, E y K
- Su proveedor puede aconsejar otros tratamientos si usted tiene heces muy duras
Ivacaftor, lumacaflor, tezacaftor y elexacaftor son medicamentos que tratan ciertos tipos de FQ.
- Mejoran la función de uno de los genes defectuosos que causan la FQ.
- Hasta el 90% de los pacientes con FQ y elegibles para usar uno o más de estos medicamentos solos o en combinación.
- Como resultado, hay menos acumulación de moco espeso en los pulmones. Otros síntomas de la FQ también mejoran.
Las medidas de cuidados personales y monitoreo en el hogar deben incluir:
- Evitar el humo, el polvo, la suciedad, los vapores, los químicos de uso doméstico, el humo de la chimenea y el moho o los hongos.
- Darles bastantes líquidos, especialmente a los bebés y niños, en clima cálido, cuando hay diarrea o heces sueltas, o durante la actividad física extra.
- Hacer ejercicio 2 o 3 veces por semana. Nadar, trotar y montar en bicicleta son buenas opciones.
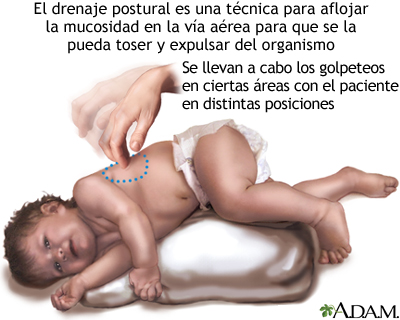
- Evacuar o sacar el moco o las secreciones de las vías respiratorias. Esto debe hacerse de 1 a 4 veces todos los días. Los pacientes, las familias y los cuidadores deben aprender a realizar la percusión torácica y el drenaje postural para ayudar a mantener las vías respiratorias despejadas.
- No se recomienda el contacto con otras personas con FQ, ya que pueden intercambiar infecciones (esto no es válido para los miembros de la familia).
Grupos de apoyo
Usted puede aliviar el estrés causado por la enfermedad formando parte de un
Expectativas (pronóstico)
La mayoría de los niños con FQ permanecen con buena salud hasta que llegan a la adultez. Pueden participar en la mayoría de las actividades y asistir a la escuela. Muchos adultos jóvenes con FQ terminan la universidad o encuentran empleo.
Con el tiempo, la enfermedad pulmonar empeora al punto en que la persona queda incapacitada. Actualmente, el período de vida promedio de las personas que padecen esta enfermedad y que viven hasta la adultez es de aproximadamente 44 años.
La muerte casi siempre es ocasionada por complicaciones pulmonares.
Posibles complicaciones
La complicación más común es la infección respiratoria crónica.
Otras complicaciones abarcan:
- Problemas intestinales, como los cálculos biliares, la obstrucción intestinal y el prolapso rectal
- Expectoración con sangre
- Insuficiencia respiratoria crónica
- Diabetes
- Esterilidad
- Enfermedad del hígado o insuficiencia hepática, pancreatitis, cirrosis biliar
- Desnutrición
- Sinusitis y pólipos nasales
- Osteoporosis y artritis
- Neumonía que continúa reapareciendo
- Neumotórax
- Insuficiencia cardíaca del lado derecho (cor pulmonale)
- Cáncer de colorrectal
Cuándo contactar a un profesional médico
Llame a su proveedor si un bebé o un niño tiene síntomas de FQ y presentan:
- Fiebre, aumento de la tos, cambios en el esputo o sangre en el esputo, inapetencia u otros signos de neumonía
- Incremento de la pérdida de peso
- Deposiciones o heces más frecuentes que tienen mal olor o tienen más mucosidad
- Abdomen hinchado o aumento de la distensión
Llame a su proveedor si alguna persona con FQ presenta síntomas nuevos o si los síntomas empeoran, particularmente
Prevención
La FQ no puede prevenirse. El hecho de realizar pruebas de detección en aquellas personas con antecedentes familiares de esta enfermedad puede detectar el gen de la FQ en muchos portadores.
Puntos de atención
Referencias
Donaldson SH, Pilewski JM, Griese M, et al. Tezacaftor/ivacaftor in subjects with cystic fibrosis and F508del/F508del-CFTR or F508del/G551D-CFTR. Am J Respir Crit Care Med. 2018;197(2):214-224. PMID: 28930490
Eagan ME, Schechter MS, Voynow JA. Cystic fibrosis. In: Kliegman RM, St. Geme JW, Blum NJ, Shah SS, Tasker RC, Wilson KM, eds. Nelson Textbook of Pediatrics. 21st ed. Philadelphia, PA: Elsevier; 2020:chap 432.
Farrell PM, White TB, Ren CL, et al. Diagnosis of cystic fibrosis: consensus guidelines from the Cystic Fibrosis Foundation. J Pediatr. 2017;181S:S4-S15.e1. PMID: 28129811
Graeber SY, Dopfer C, Naehrlich L, et al. Effects of lumacaftor/ivacaftor therapy on CFTR function in Phe508del homozygous patients with cystic fibrosis. Am J Respir Crit Care Med. 2018;197(11):1433-1442. PMID: 29327948
Grasemann H. Cystic fibrosis. In: Goldman L, Schafer AI, eds. Goldman-Cecil Medicine. 26th ed. Philadelphia, PA: Elsevier; 2020:chap 83.
Solomon GM, Hoover W, Sorscher EJ, Rowe SM. Cystic fibrosis: diagnosis and management. In: Broaddus VC, Ernst JD, King TE, Lazarus SC , Sarmiento KF, Schnapp LM, Stapleton RD, eds. Murray and Nadel's Textbook of Respiratory Medicine. 7th ed. Philadelphia, PA: Elsevier; 2022:chap 68.
Taylor-Cousar JL, Munck A, McKone EF, et al. Tezacaftor-ivacaftor in patients with cystic fibrosis homozygous for phe508del. N Engl J Med. 2017;377(21):2013-2023. PMID: 29099344
Actualizado: jueves 24 de febrero de 2022
Versión en inglés revisada por: Neil K. Kaneshiro, MD, MHA, Clinical Professor of Pediatrics, University of Washington School of Medicine, Seattle, WA. Also reviewed by David Zieve, MD, MHA, Medical Director, Brenda Conaway, Editorial Director, and the A.D.A.M. Editorial team.
Traducción y localización realizada por: HolaDoctor, Inc.
La información aquí contenida no debe utilizarse durante ninguna emergencia médica, ni para el diagnóstico o tratamiento de alguna condición médica. Debe consultarse a un médico con licencia para el diagnóstico y tratamiento de todas y cada una de las condiciones médicas. En caso de una emergencia médica, llame al 911. Los enlaces a otros sitios se proporcionan sólo con fines de información, no significa que se les apruebe. © 1997-2025 A.D.A.M., Inc. La reproducción o distribución parcial o total de la información aquí contenida está terminantemente prohibida.